Working in the context of computational chemistry, I focus on method development for fast yet reliable total energy estimations. In my view, it is key to include as many possible applications in the design of the methods in order to avoid having convoluted hard-to-use methods tailored only to an individual problem. This is why I enjoy including concepts from a range of my previous work (global cluster optimisation, large-scale classical molecular dynamics, and development for Density Functional Theory code) in current projects.
Currently, I am affiliated with the group of Prof. von Lilienfeld at University of Basel working on predictions of total electronic energies based on perturbation of a molecular core potential or machine learning.
Quantum Alchemy
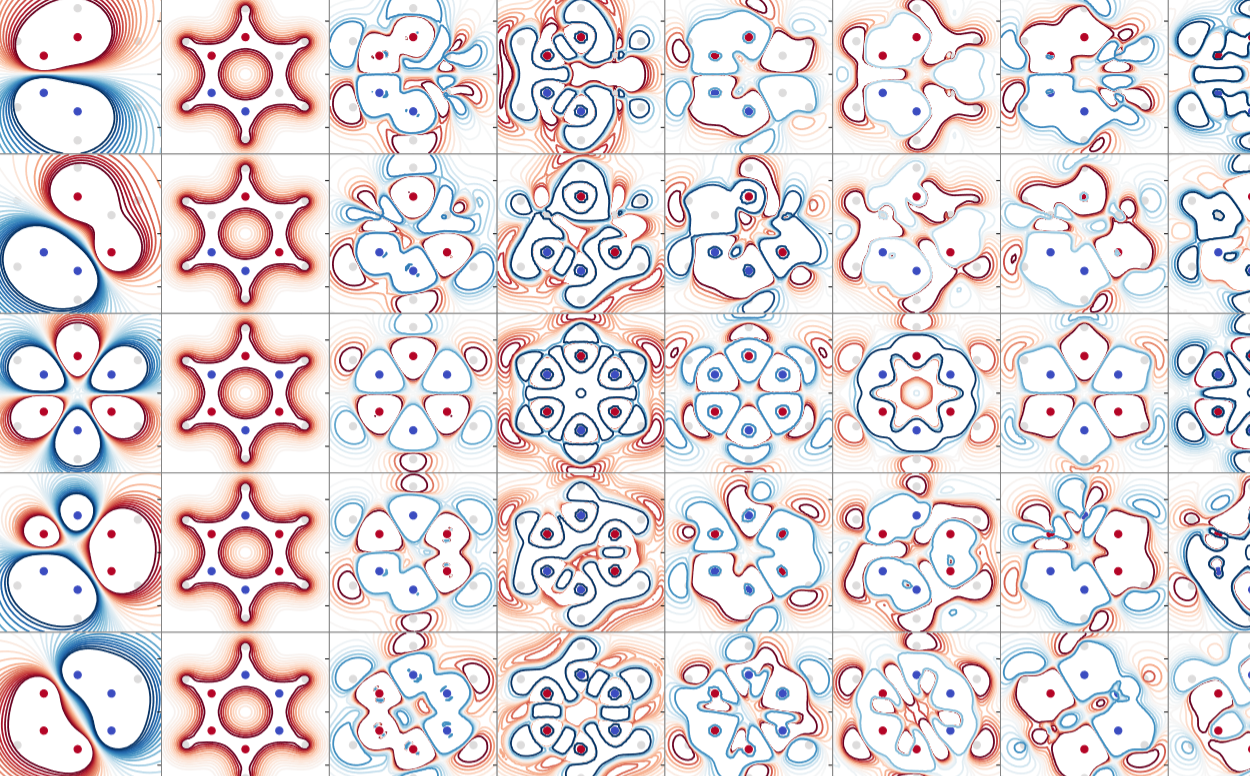
I develop Quantum Alchemy for materials design problems to assess millions of molecules at once instead of scanning them one-by-one.
Solid/liquid Interfaces
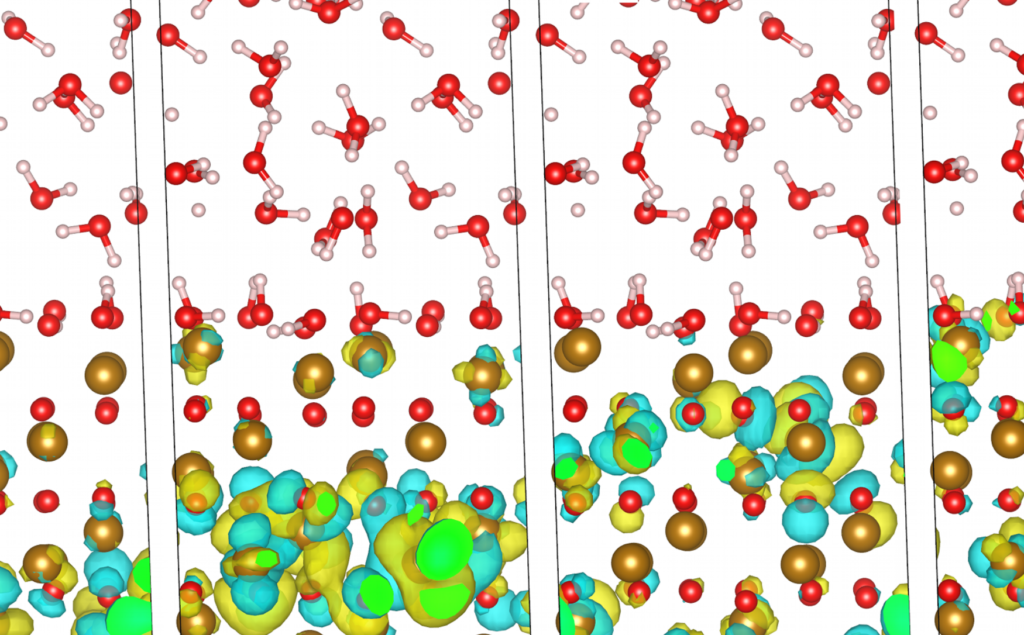
Solid/liquid interfaces are at the core of many applications. I work on describing protonation states and surface reorganisation.
Machine Learning Models
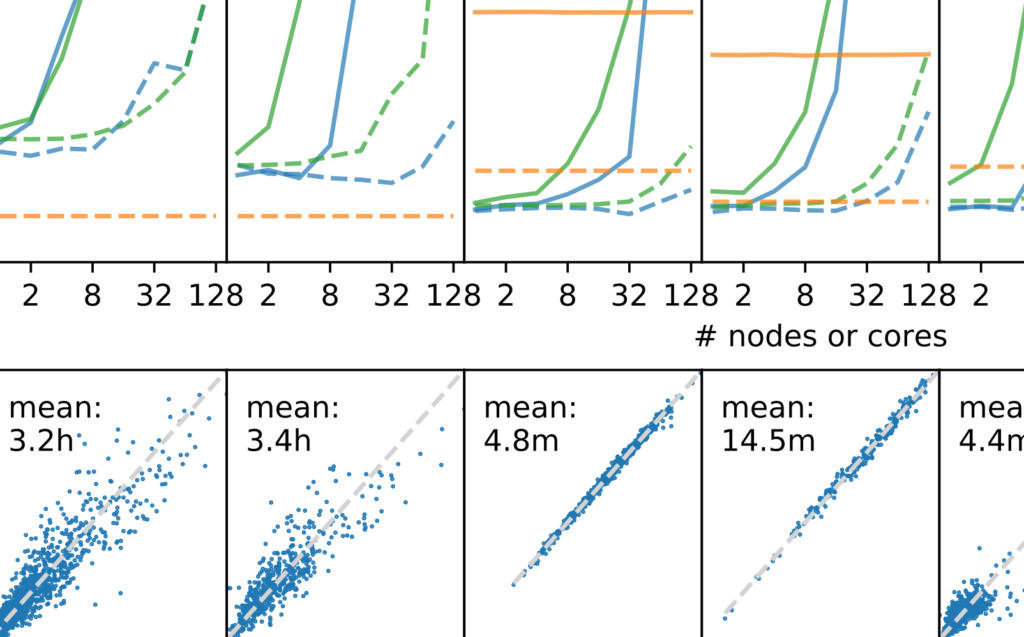
Non-parametric models are powerful tools. I use them to model reaction barriers and scheduling problems.
Past projects:
- Fluorinated organic compounds in membrane environments, Prof. Sebastiani, Martin-Luther-Universität Halle
- Global optimisation algorithms for atomic potentials, Prof. Sebastiani, Freie Universität Berlin