
If you do unrestricted DFT calculations with CP2K for charged systems, you may wonder which spin channel the electron is taken from or added to. Moreover, the sign convention for any output (Hirshfeld or Mulliken spin charge, cube files) may be of interest to you.
computational chemistry
34 posts
This is an ongoing effort to collect error messages from CP2K which typically cannot be understood as long as one has a look into the source code. You have not found the error that is bugging you on this page? Try also part 1.

In spectroscopy and related fields, a notation for states is used that holds a lot of symmetry information to support employing selection rules. The only problem I have is that I use this to rarely to remember all the meanings. Moreover, this notation is exceptionally hard to resolve using search […]
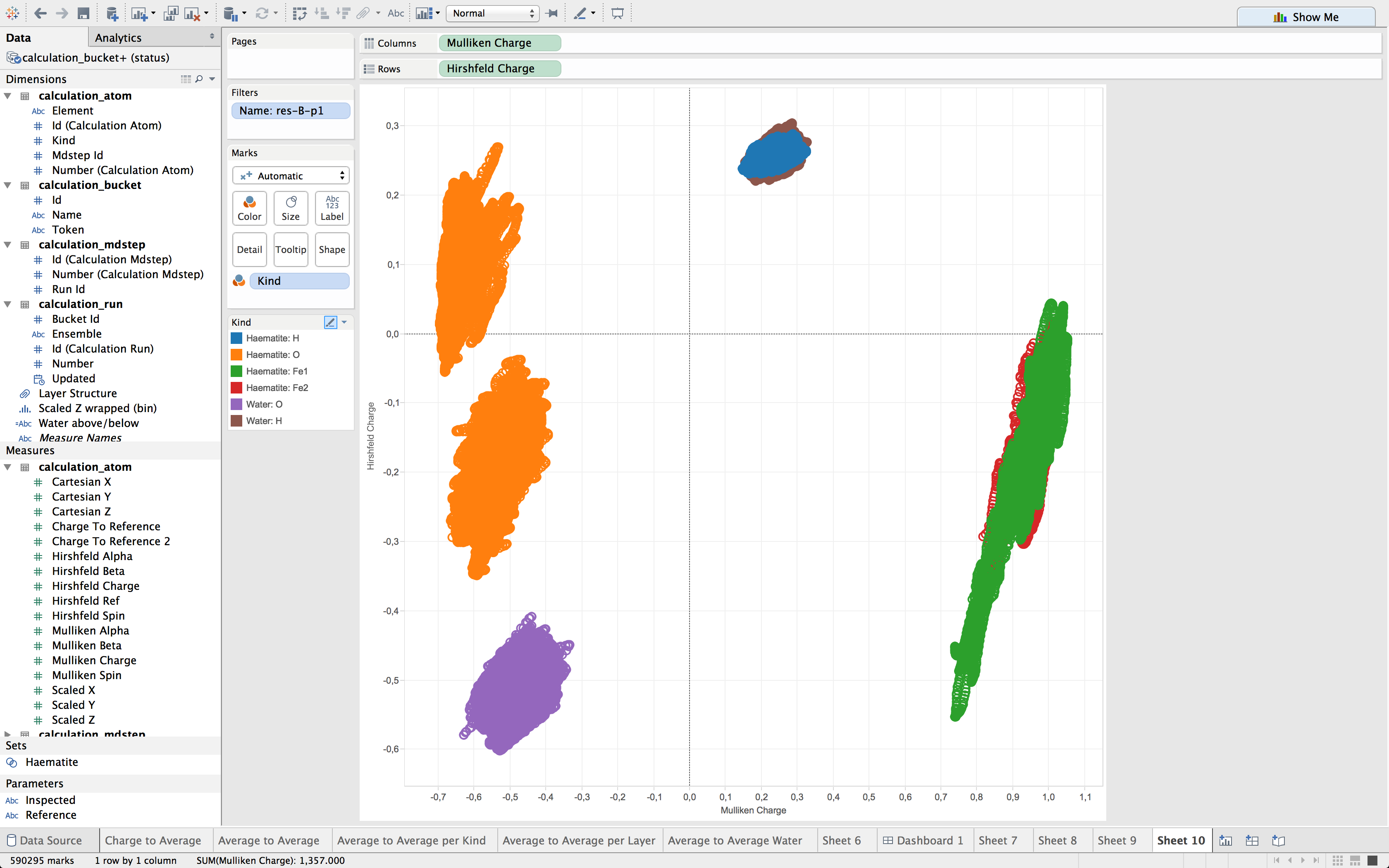
A few weeks ago, I came across Tableau which can be useful in my field of research despite the fact that it is meant to be used for something completely different.
Calculating the hydrogen bond occupancy is a frequent task in the analysis of molecular dynamics simulations. Therefore, it has been implemented in packages like VMD hbonds or MDAnalysis.analysis.hbonds. However, these implementations do not fully treat periodic boundary conditions, as hydrogen bonds crossing the simulation box boundary are not incorporated into […]
Sometimes, it is helpful to get the index (number starting from zero) or serial (number starting from one) for the currently drawn part of the molecule in vmd. For example, it you want to create an index file for a given system, or if you want to split up your […]
Sometimes, one has the bond topology but not the matching angle or dihedral definitions. Here is how to get them in VMD with topotools.
This is an ongoing effort to collect error messages from CP2K which typically cannot be understood as long as one has a look into the source code. If you have not found your problem here, try also part 2 of this list.

Research is a widely understood as a collaborative effort. This graphic illustrates how researchers from all over the world work together towards a single objective: better understanding of nature. I selected a small community that is working on surface physics of hematite, a mineral that is particularly useful as cost-effective […]
Embedding one molecule (e.g., a protein) into another one (e.g., a membrane) has to be done carefully in order to avoid any conformational clashes. For the GROMACS world, there is g_membed, but for namd there is no tool that I know of. So here is a TCL script to help […]
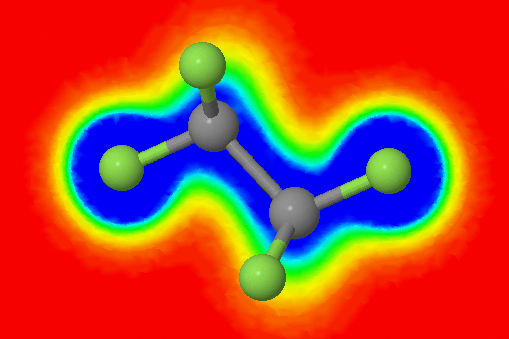
This short article will guide you through the creation of images like the one above based on charge density calculations using Gaussian09. For visualization, I will use Jmol. The principal steps are: Get a conformation of the molecule you are interested in. Optimize this geometry until you find a local […]